The focus of software development in the Hammell lab revolves around improving quality control and maximizing data recovery from high throughput sequencing projects. In particular, the lab is interested in improving bioinformatics analysis of repetitive sequences, particularly transposable elements, in order to elucidate novel (and previously ignored) biological insights of their functions in development and diseases.
We have developed the following tools to address various bioinformatics needs:
TEToolkit suite
- TEtranscripts: A package for including transposable elements in differential enrichment analysis of sequencing datasets.
- TEsmall: A package for including small RNAs derived from repetitive regions in differential enrichment analysis.
- TEpeaks: A package for including repetitive regions in peak calling from ChIP-seq datasets.
- TElocal: A package for quantifying transposable elements at a locus level of RNA-seq datasets.
Other software
Choosing the right software
Handling transposon derived reads can be tricky with short read sequencing data. There are now lots of software packages out there, but how do you pick the right one for your needs?
In this review, we describe all of the software that’s been developed for handling transposable elements in common genomics assays, such as: RNA-seq, ChIP-seq, CLIP-seq, DNA methyl-seq, and small RNA data. There’s also lots of room for improvement, both computationally and experimentally.
We hope this review will be helpful both to TE experts as well as newcomers to the transposon field.
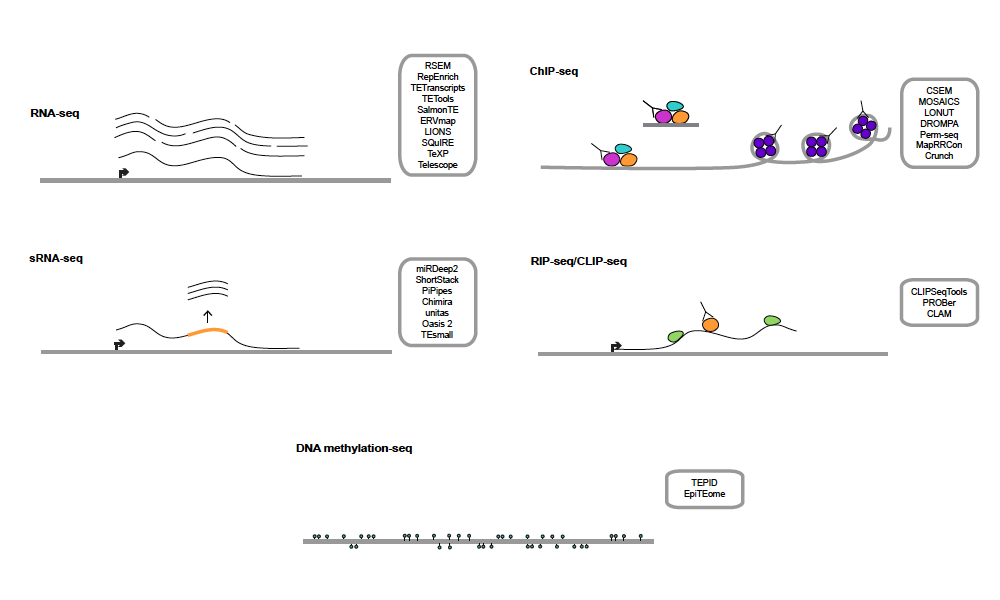
Summary
TEtranscripts is a software package that utilizes both unambiguously (uniquely) and ambiguously (multi-) mapped reads to perform differential enrichment analyses from high throughput sequencing experiments. Currently, most expression analysis software packates are not optimized for handling the complexities involved in quantifying highly repetitive regions of the genome, especially transposable elements (TE), from short sequencing reads. Although transposon elements make up between 20 to 80% of many eukaryotic genomes and contribute significantly to the cellular transcriptome output, the difficulty in quantifying their abundances from high throughput sequencing experiments has led them to be largely ignored in most studies. The TEtranscripts provides a noticeable improvement in the recovery of TE transcripts from RNA-Seq experiments and identification of peaks associated with repetitive regions of the genome.
If you encounter any issues or have any questions about TEtranscripts, please refer to our FAQ page, or check out our Github page.
Download instructions
You can download the software package from PyPi and GitHub. The transposable element GTF files required by TEtranscripts (see tool description below) and example data files (BAM) are available at this location. GTF files can also be downloaded here.
Tool Description
The two tools, TEtranscripts and TEcount, quantify both gene and transposable element (TE) transcript abundances from RNA-Seq experiments, utilizing both uniquely and ambiguously mapped short read sequences. It processes the short reads alignments (BAM files) and proportionally assigns read counts to the corresponding gene or TE based on the user-provided annotation files (GTF files). In addition, TEtranscripts combines multiple libraries and perform differential analysis using DESeq2.
GTF files for gene annotation can be obtained from UCSC RefSeq, Ensembl, iGenomes or other annotation databases. GTF files for TE annotations are customly generated from UCSC RepeatMasker or other annotation database. They contain two custom attributes, class_id and family_id, corresponding to the class (e.g. LINE) and family (e.g. L1) of the corresponding transposable element. A unique ID (e.g. L1Md_Gf_dup1) is also assigned for each TE annotation in the transcript_id attribute. Pre-generated TE GTF files are available for a number of organisms, and can be downloaded here or here. If the organism or genome build of your interest is not available, please contact us and provide a curated annotation of the transposable elements (e.g. genomic location and TE name/type). We will do our best to help you generate the suitable TE GTF file.
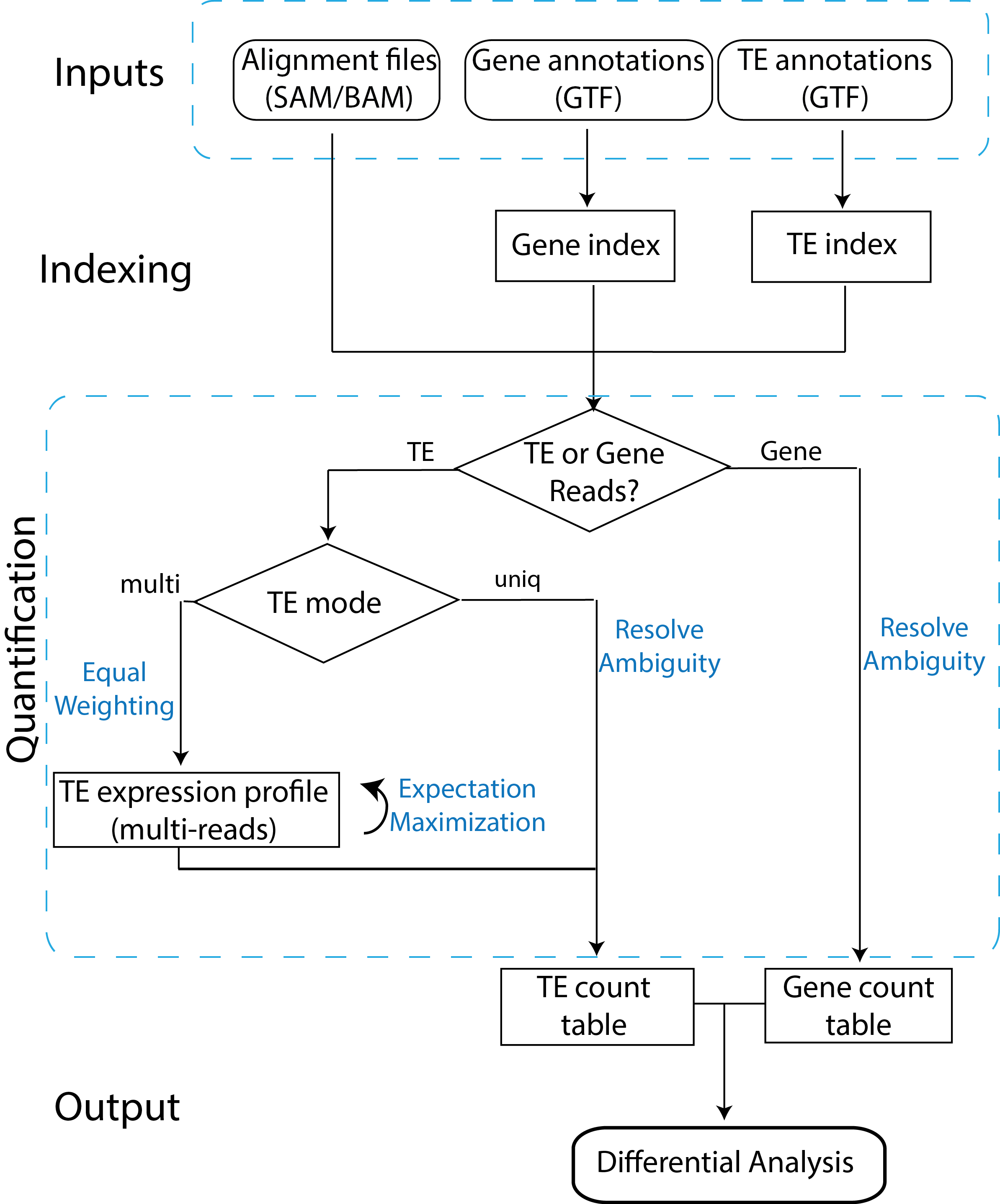
TEtranscripts analysis workflow
The TEtranscripts software package is written for Python (2.6.x or 2.7.x), and requires pysam (v0.9.x or higher), R (2.15.x or greater) and DESeq2 (1.10.x or greater).
TEtranscripts is an open-source software released under the GNU General Public License version 3 (GPLv3).
Citation
Please cite the following article when using TEtranscripts:
- Jin Y., Tam O.H., Paniagua E. and Hammell M. (2015). TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31: 3593-3599. Pubmed ID: 26206304
For more information about how to use TEtranscripts:
- Jin, Y. and Hammell, M. (2018) Analysis of RNA-Seq Data Using TEtranscripts. Methods Mol Biol, 1751: 153-167. Pubmed ID:29508296
Summary
TEsmall is a tool that allows for the simultaneous processing and analysis of a variety of small RNAs in a single integrated workflow. These include microRNA (miRNA), transfer RNA (tRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), Y-RNA, Piwi-interacting RNA (piRNA), and short interfering RNA (siRNA). This package facilitates the discovery of intriguing biological phenomena otherwise masked by insufficient annotation of repetitive genomic elements, such as siRNAs, and allows these elements to be easily incorporated into downstream differential analysis through packages like DESeq2.
If you encounter any issues or have any questions about TEsmall, please check out our Github page.
Download instructions
You can download the software package from GitHub, with detailed instructions on installation using Miniconda. Additional packages required by the TEsmall workflow include cutadapt, bowtie, bedtools, samtools, pybedtools, and scipy.
Tool Description
TEsmall functions by accepting raw input in FASTQ file format from next generation sequencing platforms in conjunction with genomic annotation sets via an online server. Adapters are trimmed from FASTQ reads by the cutadapt package, and rRNA derived reads are next filtered from the data by aligning to rRNA sequences using bowtie. Filtered reads are then aligned to the genome using bowtie, allowing no mismatches and up to 100 alignments (reads with more alignments are discarded). The choice of 100 genomic loci as the upper limit allows for the classification of multimapper reads common to sRNA data, in particular structural RNAs like tRNAs and transposable element targeting siRNAs, while removing or reducing homopolymer or low complexity reads from downstream analysis.
Following alignment to the genome, each alignment is annotated via a sequential decision tree, where each alignment are distributed to an annotation category in order, then removed from the pool of alignments in order to facilitate priority annotation. The default order/priority of annotation is: structural RNAs, miRNAs and hairpins, exons, sense transposons, antisense transposons, introns, and ultimately annotated piRNA clusters. This annotation class priority can be re-ordered by the user to suit the application and user preferences.
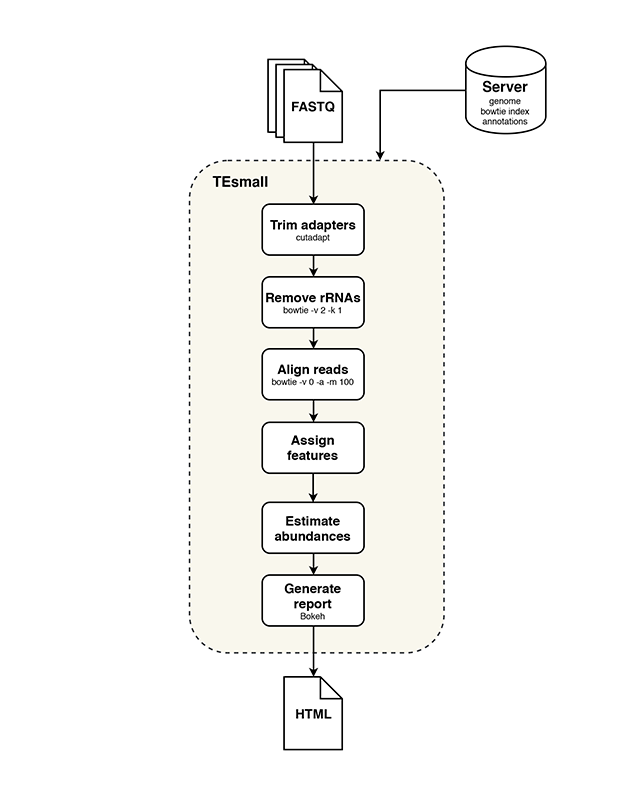
TEsmall workflow
An HTML output file is then created using python based Bokeh tools to visualize the abundance distributions, length distributions, and mapping logs of all small RNAs in the dataset. In conjunction with this HTML output, TEsmall compiles multiple flat text output files, including a counts file that is structured to be directly compatible with DESeq2 for differential analysis. The abundance calculations for these counts files are 1/n normalized at the end of this annotation process, where n represents the number of alignments per read, to ensure no double-counting of multimappers.
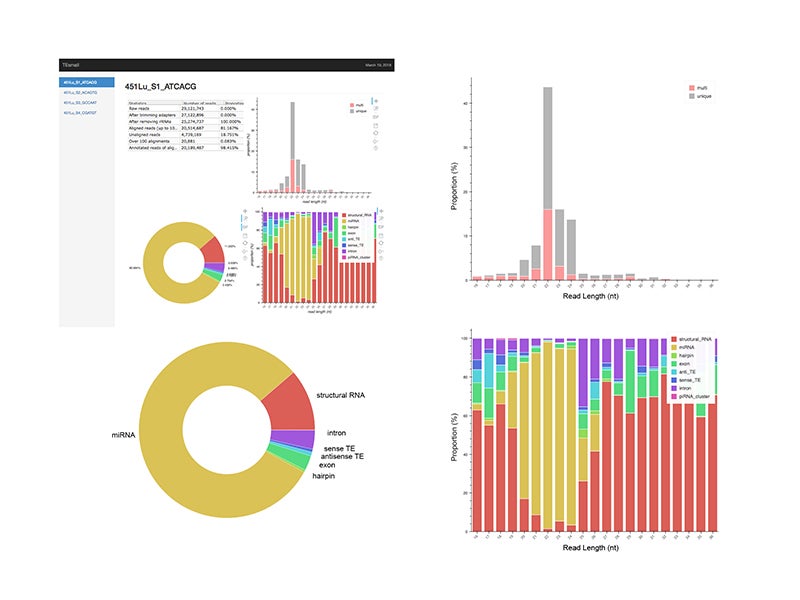
TEsmall output
Annotation files
Annotation files for the human (hg19), mouse (mm9) and fly (dm3) genomes can be found here. For other genome builds, you would need the following files
- Sequences
- FASTA file of genomic sequences (genome.fa), with a corresponding index generated by
samtools faidx - FASTA file of rDNA sequences (rDNA.fa), with a corresponding index generated by
samtools faidx - Bowtie indices (
bowtie-build) for the genomic sequence - Bowtie indices (
bowtie-build) for the rDNA sequences
- FASTA file of genomic sequences (genome.fa), with a corresponding index generated by
- BED files for annotation
- Gene exons (exon.bed)
- Gene introns (intron.bed)
Transposable elements (TE.bed)
- Mature microRNA sequence (miRNA.bed)
- MicroRNA hairpin (hairpin.bed)
- PIWI-interacting RNA clusters (piRNA_cluster.bed)
- Other small non-coding RNA, such as tRNA, snRNA (structural_RNA.bed)
Empty BED files must be provided even if there are no applicable annotations for a particular category.
If you are encountering difficulty generating the files for your genome build, you can contact us, and we will help you to the best of our abilities.

Annotation workflow
The TEsmall software package is written for Python (2.x or 3.x).
TEsmall is an open-source software released under the GNU General Public License version 3 (GPLv3).
Please cite the following article when using TEsmall:
- O’Neill K., Liao W.W., Patel A. and Gale-Hammell M. (2018) TEsmall Identifies Small RNAs Associated With Targeted Inhibitor Resistance in Melanoma. Front. Genet. 9: 461. Pubmed ID: 30349559
Summary
SAKE (Single-cell RNA-seq Analysis and Klustering Evaluation) is a robust method for single cell RNA-seq analysis that provides quantitative statistical metrics at each step of the analysis pipeline. This toold provides several modules that include: data pre-processing for quality control, sample clustering, t-SNE visualization of clusters, differential expression between clusters, and functional enrichment analysis. The aim of SAKE is to provide a user-friendly tool for easy analysis of NGS Single-Cell transcriptomic data.
If you encounter any issues or have any questions about SAKE, please check out the Github page
Download instructions
You can download the software package from GitHub, with detailed installation instructions and a full list of prerequisite libraries.
Tool Description
SAKE workflow is designed to robustly categorize gene expression profiles while avoiding unwanted noise. It utilizes a table of estimated gene abundances as input, and begins by removing samples with low total transcript counts and gene coverage. Low abundance transcripts are then filtered using median absolute deviation (MAD) to reduce stochastic dropout and technical noise. Clustering is then performed using Non-negative Matrix Factorization (NMF) with a variety of k values (corresponding to the number of clusters) and produces visual representation of the results to identify the optimal k value. After identifying the optimal cluster number, SAKE performs a large number of iterations with NMF to identify robust sample and marker membership to each cluster. The clustering results can then be visualized on t-SNE, and differential analyses and enrichment analyses can be performed to further characterize the identified clusters.
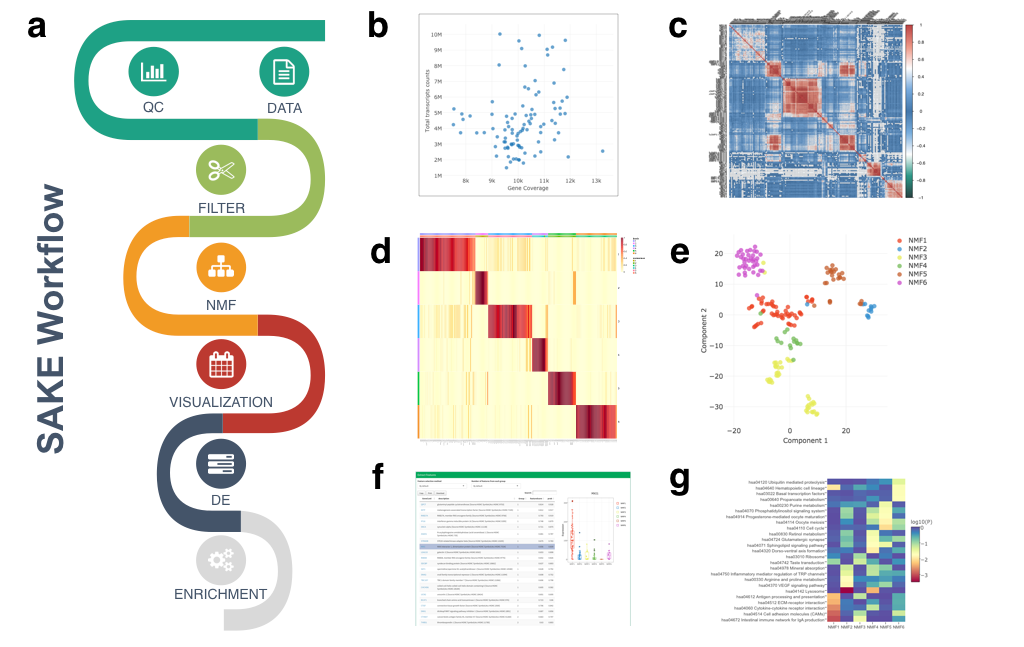
Flowchart of SAKE package and example analysis results: a) Analysis workflow for analyzing single-cell RNA-Seq data. b) Quality Controls to compare total sequenced reads to total gene transcripts detected. c) Sample correlation heat map plot d) A heat map of sample assignment from NMF run, with dark red indicating high confidence in cluster assignments e) t-SNE plot to compare NMF assigned groups with t-SNE projections. f) A table of NMF identified features (genes defining each cluster) and a box plot of gene expression distributions across NMF assigned groups. g) Summary table for GO term enrichment analysis for each NMF assigned group.
Please cite the following article when using SAKE:
- Ho Y.J., Anaparthy N., Molik D., Mathew G., Aicher T., Patel A., Hicks J. and Gale Hammell M. (2018) Single-cell RNA-seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res. 28: 1353-1363. Pubmed ID: 30061114
Summary
ezBAMQC is a tool to check the quality of either one or many mapped next-generation-sequencing datasets. It conducts comprehensive evaluations of aligned sequencing data from multiple aspects including: clipping profile, mapping quality distribution, mapped read length distribution, genomic/transcriptomic mapping distribution, inner distance distribution (for paired-end reads), ribosomal RNA contamination, transcript 5’ and 3’ end bias, transcription dropout rate, sample correlations, sample reproducibility, sample variations. It outputs a set of tables and plots and one HTML page that contains a summary of the results. Many metrics are designed for RNA-seq data specifically, but ezBAMQC can be applied to any mapped sequencing dataset such as RNA-seq, CLIP-seq, GRO-seq, ChIP-seq, DNA-seq and so on.
If you encounter any issues or have any questions about ezBAMQC, please contact us or check out our Github page
Download instructions
You can download the source code from PyPi or GitHub, or a pre-compiled version of the software package from GitHub.
Tool Description
The ezBAMQC software package is written for Python 2.7.x. To install ezBAMQC from a pre-compiled package, it requires pysam (v0.8.3 or higher), R (2.15.x or greater) and the corrplot R package. If compiling ezBAMQC from source, you will need a compiler with C++11 support. This is provided by GNU GCC (version 4.8.1 or greater) in Linux, or Xcode (version 4.2 or greater) in MacOSX.
ezBAMQC is an open-source software released under the GNU General Public License version 3 (GPLv3).
Note: ezBAMQC will not work with Python 3.
Citation
Please see our Github page for more details
Summary
TEpeaks is a tool to perform peak-calling of ChIP-seq datasets using both uniquely mapped and multi-mapped (ambiguous) reads.
Download instructions
You can download the source code from GitHub.
Tool Description
Please contact us for more details
Citation
Please see our Github page for more details
Summary
TElocal is a tool that utilizes both uniquely and ambiguously mapped reads to quantify transposable element expression at the locus level.
If you encounter any issues or have any questions about TElocal, please refer to our Github page.
Download instructions
You can download the source code from GitHub. The pre-built transposable element GTF indices required by TElocal are available at this location. The genomic position of the transposable elements in the pre-built GTF indices are available here.
Tool Description
Please see our README on Github for more information.
Citation
Please see our Github page for more details